HPC
In this session we will look at running code on compute clusters.
An initial consideration is always going to be 'when should I run code on the cluster rather than my laptop?'. I think the following are the main reasons:
- Not enough resources on your machine (e.g. not enough memory, needs a high-end GPU).
- Job will take longer than you want to leave you machine on for.
- Running many jobs in parallel.
- Using data which is more easily/only accessed through cluster storage.
- A software installation which works more easily on cluster OS (e.g. linux rather than OS X).
- Benchmarking code.
Of course it's almost always easier to run code on your local machine if you can avoid these cases. Weighing up effort to run on the HPC versus the effort of finding workarounds to run locally is hard. Making the HPC as painless as possible, while acknowledging there will always be some pain, is probably the best approach.
Particular pain-points of using the HPC can be:
- Clunky interface, terminal only.
- Waiting for your jobs to run.
- Running jobs interactively.
- Installing software.
- Troubleshooting jobs or getting code to run.
- Running many jobs at once and resubmitting failed jobs.
- Making your workflow reproducible.
I've tried to present solutions to these in turn, including three exercises:
- Estimating resource use.
- Installing software without admin rights.
- Running a job array.
Some of this will be specific to the current setup of the EBI codon cluster. I'm sticking with LSF for now -- a change to SLURM is planned because apparently it has better metrics centrally. Similar principles apply as with LSF, some features do change. For this year cloud computing won't be covered (sorry), but I can try and add this in future. So the topics for today:
- Maximising your quality of life on the HPC
- LSF basics (recap)
- Getting jobs to run and requesting resources
- Interactive jobs
- Installing software without admin rights
- Troubleshooting jobs and prototyping code
- Job arrays
- Reproducibility on the HPC
Maximising your quality of life on the HPC
There are some basic tools you can use for bash/zsh and Unix which will make your time on the HPC less unpleasant. I'll mention them here -- if they look useful try them out after the class.
If anyone has further suggestions, now is a good time to discuss them.
tmux
This is my biggest piece of advice in this section: always use tmux to maintain
your session between log ins. (You may have used screen before, tmux has more
features and is universally available).
The basic usage is:
- When you log in for the first time, type
tmux new(ortmux new zshif you wanted to run a specific terminal). - This is your session, run commands as normal within the interface.
- When it's time to log out, press
Ctrl-bthend(for detach). - Next time you log in, type
tmux attachto return to where you were. - Use
Ctrl-bthencto make a new tab.Ctrl-band a number or arrow to switch between. - Use
Ctrl-bthen[then arrows to scroll (but easier to make this the mouse as normal through config file).
If you crash/lose connection the session will still be retained. Sessions are lost
when the node reboots. Also note, you need to specify a specific node e.g. codon-login-01
rather than the load-balancer codon-login when using tmux.
There are many more features available if you edit the ~/.tmux.conf file. In particular
it's useful to turn the mouse on so you can click between panes and scroll as normal.
Here's an old one of mine with more
examples.
Background processes
Less useful on a cluster, but can be if you ever use a high-performance machine without a queuing system (e.g. openstack, cloud).
Ctrl-z suspends a process and takes you back to the terminal. fg to foreground (run
interactively), bg to keep running but keep you at the prompt. See jobs to look
at your processes.
You can add an ampersand & at the end of a command and it will run in the background
without needing to suspend first. nohup will keep running even when you log out
(not necessary if you are using tmux).
Text editors
Pick one of vi/vim/gvim/neovim or emacs and learn some of the basic commands.
Copying and pasting text, find and replace, vertical editing and repeating custom commands
are all really useful to know. Once you're used to them, the extensions for VSCode which
let you use their commands are very powerful.
Although, in my opinion, they don't replace a modern IDE such as VSCode every system has them installed and it's very useful to be able to make small edits to your code or other files. Graphical versions are also available.
Terminal emulators
Impress your friends and make your enemies jealous by using a non-default terminal emulator. Good ones:
zsh and oh-my-zsh
I personally prefer zsh over bash because:
- History is better. Type the start of a command, then press up and you'll get the commands you've previously used that started like that. For example, type
lsthen press up. - The command search
Ctrl-Ris also better. - Keep pressing tab and you'll see options for autocomplete.
I'm sure there are many more things possible.
zsh is now the default on OS X. You can change shell with chsh, but you usually
need admin rights. On the cluster, you can just run zsh to start a new session,
but if you're using tmux you'll pop right back into the session anyway.
You can also install oh-my-zsh which has some nice extensions, particularly for within git directories. The little arrow shows you whether the last command exited with success or not.
Enter-Enter-Tilde-Dot
This command exits a broken ssh session. See here for more ssh commands.
Other handy tools I like
agrather thangrep.fdrather thanfind.fzffor searching through commands. Can be integrated withzsh.
While I'm writing these out, can't resist noting one of my favourite OS X commands:
xattr -d "com.apple.quarantine" /Applications/Visual\ Studio\ Code.app
Which gets rid of the stupid lock by default on most executables on OS X.
LSF basics (recap)
- Submit jobs with
bsub. - Check your jobs with
bjobs.bjobs -dto include recently finished jobs. - View the queues with
bqueues. Change jobs withbswitch. bstop,bresume,brestartto manage running jobs. End jobs withbkill.bhostsandlshoststo see worker nodes.bmodto change a job.
Most of these you can add -l to get much more information. bqueues -l is interesting
as it shows your current fair-share priority (lower if you've just run a lot of jobs).
There are more, but these are the main ones. Checkpointing doesn't work well, as far as I know.
Getting jobs to run and requesting resources
It's frustrating when your jobs take a long time to run. Requesting the right amount of resources both helps your job find a slot in the cluster to run, and lowers the load overall helping everyone's jobs to run.
That being said, I don't want to put anyone off experimenting or running code on the HPC. Don't worry about trying things out or if you get resource use wrong. Over time improving your accuracy is worthwhile, but it's always difficult with a new task -- very common in research! We now have some nice diagnostics and history here I'd recommend checking out periodically to keep track of how you're doing: http://hoxa.windows.ebi.ac.uk:8080/.
The basic resources for each CPU job are:
- CPU time (walltime)
- Number of CPU cores
- Memory
/tmpspace
Though the last of these isn't usually a concern, we have lots of fast scratch space you can point to if needed. Disk quotas are managed outside of LSF and I won't discuss them here.
When choosing how much of these resources to request, you want to avoid:
- The job failing due to running out of resources.
- Using far less than the requested resources.
- Long running jobs.
- Inefficient resource use (e.g. partial parallelisation, peaky maximum memory).
- Requesting resources which are hard to obtain (many cores, long run times, lots of memory, lots of GPUs).
- Lots of small jobs.
- Inefficient code.
Typically 1) is worse than 2). Those resources were totally wasted rather than partially wasted. We will look at this in the exercise.
Long running jobs are bad because failure comes at a higher cost, and it makes the cluster use less able to respond to dynamic demand. Considering adding parallelisation rather than a long serial job if possible, or adding some sort of saving of state so jobs can be resumed.
You can't usually do much about the root causes of 4) unless you wrote the code yourself. You will always need a memory request which is at least the maximum demand (you can have resource use which changes over the job, but it's not worth it unless e.g. you are planning the analysis of a biobank). Inefficient parallel code on a cluster may be better as a serial job. Note that if you don't care about the code finishing quickly it's always more efficient in CPU terms to run them on a single core. For example, if you have 1000 samples which you wished to run genome assembly on would you run:
- 1000 single-threaded jobs
- 1000 jobs with four threads
- 1000 jobs with 32 threads?
(we will discuss this)
-
can sometimes be approached by using job arrays or trading off your requests. Sometimes you're just stuffed.
-
is bad because of the overhead of scheduling them. Either write a wrapper script to run them in a loop, or use the
shortqueue. -
at some point it's going to be worth turning your quickly written but slow code into something more efficient, as per the previous session. Another important effect can come from using the right compiler optimisations, which we will discuss next week.
Anatomy of a bsub
Before the exercise, let's recap a typical bsub command:
bsub -R "select[mem>10000] rusage[mem=10000]" -M10000 \
-n8 -R "span[hosts=1]" \
-o refine.%J.o -e refine.%J.e \
python refine.py
-R "select[mem>10000] rusage[mem=10000]" -M10000requests ~10Gb memory.-n8 -R "span[hosts=1]"requests 8 cores on a single worker node.-o refine.%J.o -e refine.%J.ewrites STDOUT and STDERR to files named with the job number.python refine.pyis the command to run.
NB the standard memory request if you don't specify on codon is 15Gb. This is a lot, and you should typically reduce it.
If you want to capture STDOUT without the LSF header add quotes:
bsub -o refine.%J.o -e refine.%J.e "python refine.py > refine.stdout"
You can also make execution dependent on completion of other jobs with the -w flag
e.g. bsub -w DONE(25636). This can be useful in pipeline code.
When I first started using LSF I found a few commands helpful for looking through your jobs:
bjobs -a -x
find . -name "\*.o" | xargs grep -L "Successfully completed"
find . -name "\*.o" | xargs grep -l "MEMLIMIT"
find . -name "\*.e" | xargs cat
Exercise 1: Estimating resource use
You submit your job, frantically type bjobs a few times until you see it start
running, then go off and complete the intermediate programming class feedback form while you wait. When you check
again, no results and a job which failed due to running out of memory, or going over run time. What now?
You can alter your three basic resources as follows:
- CPU time: ask for more by submitting to a different queue.
- CPU time: use less wall time by asking for more cores and running more threads.
- Memory: ask for more in the
bsubcommand.
Ok, but how much more? There are three basic strategies you can use:
- Double the resource request each time.
- Run on a smaller dataset first, then estimate how much would be needed based on linear or empirical scaling.
- Analyse the computational efficiency analytically (or use someone else's analysis/calculator).
The first is a lazy but effective approach for exploring a scale you don't know the maximum of. Maybe try this first, but if after a couple of doublings your job is still failing it's probably time to gather some more information.
Let's try approach two on a real example by following these steps:
- Find the sequence alignment from last week. Start off by cutting down to just the first four sequences in the file.
- Install RAxML version 8. You can get the source here, but it's also more easily available via bioconda.
- Run a phylogenetic analysis and check the CPU and memory use. The command is of the form
raxmlHPC-SSE3 -s BIGSdb_024538_1190028856_31182.dna.aln -n raxml_1sample -m GTRGAMMA -p 1. - Increase the number of samples and retime. I would suggest a log scale, perhaps doubling the samples included each time. Plot your results.
- Use your plot to estimate what is required for the full dataset.
If you have time, you can also try experimenting with different numbers of CPU threads (use the PTHREADS version and -T), and number of sites
in the alignment too.
You can use codon or your laptop to do this.
LSF is really good at giving you resource use. What about on your laptop? Using
/usr/bin/time -v (gtime -v on OS X, install with homebrew) gives you most of the same information.
What about approach three? (view after exercise)
For this particular example, the authors have calculated how much memory is required and even have an online calculator. Try it here and compare with your results.
Adding instrumentation to your code
We said in optimisation that some measurement is better than no measurement.
I am a big fan of progress bars. They give a little more information than just the total time and are really easy to add in. When you've got a function that just seems to hang, seeing the progress and expected completion time tells you whether your code is likely to finish if you just wait a bit longer, or if you need to improve it to have any chance of finishing. This also gives you some more data, rather than just increasing the wall time repeatedly.
In python, you can wrap your iterators in tqdm which gives you a progress bar
which number of iterations done/total, and expected time.
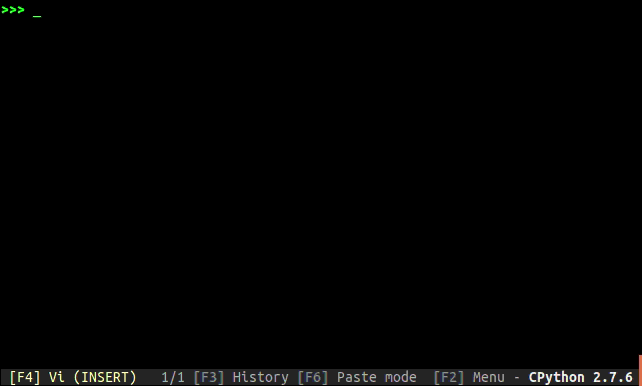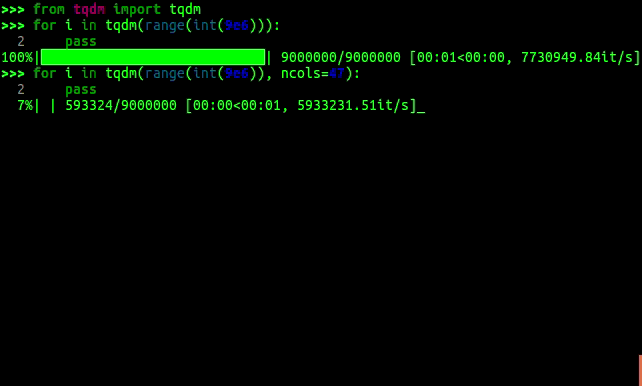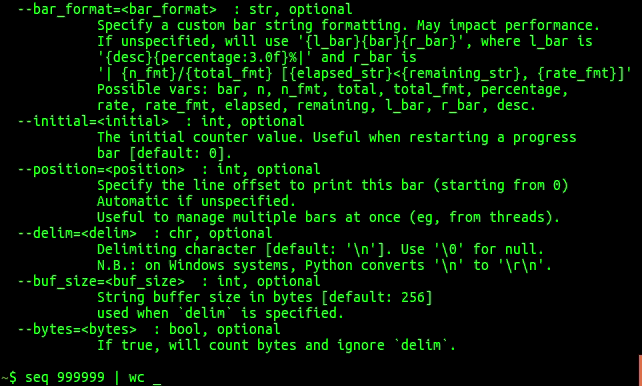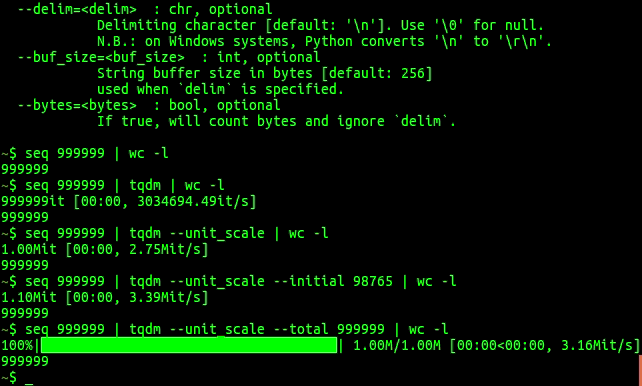
tqdm progress bars in python
GPUs
I have not used the GPUs on the codon cluster, but a few people requested that I cover them.
In the session I will take a moment if anyone has experience they would like to discuss.
The following are a list of things I think are relevant from my use of GPUs (but where I have admin rights and no queue):
- Use
nvidia-smito see available GPUs, current resource use, and driver version. - Using multiple GPUs is possible and easier if they are the same type.
- GPUs have a limited amount device memory, usually around 4Gb for a typical laptop card to 40Gb for a HPC card.
- The hierarchy of processes is: thread -> warp (32 threads) -> block (1-16 warps which share memory) -> kernel (all blocks needed to run the operation) -> graph (a set of kernels with dependencies).
- Various versions to be aware of: driver version (e.g. 410.79) -- admin managed; CUDA version (e.g. 11.3) -- can be installed centrally or locally; compute version (e.g. 8.0 or Ampere) -- a feature of the hardware. They need to match what the package was compiled for.
- When compiling, you will use
nvccto compile CUDA code and the usual C/C++ compiler for other code.nvccis used for a first linker step, the usual compiler for the usual final linker step. pytorchetc have recommended install routes, but if they don't work compiling yourself is always an option.- You can use
cuda-gdbto debug GPU code. Use is very similar togdb. Make sure to compile with the flags-g -G. - Be aware that cards have a maximum architecture version they support. This is the compute version given to the CUDA compiler as sm_XX. For A100 cards this is sm_80.
See here for some relevant installation documentation for one of our GPU packages: https://poppunk.readthedocs.io/en/latest/gpu.html
Using GPUs for development purposes? We do have some available outside of codon which are easier to play around with.
Interactive jobs
Just some brief notes for this point:
- Use
bsub -I, and without the-oand-eoptions to run your job interactively, i.e. so you are sent to the worker node terminal as the job runs. - Make sure you are using X11 forwarding to see GUIs. Connect with
ssh -X -Y <remote>, and you may also need Xquartz installed. - If you are using
tmuxyou'll need to make sure you've got the right display. Runecho $DISPLAYoutside of tmux, thenexport DISPLAY=localhost:32(with whatever$DISPLAYcontained) inside tmux. - If you want to view images on the cluster without transferring files, you can
use
display <file.png> &. For PDFs see here.
Ultimately, it's a pain, and using something like OpenStack or a VM in the cloud is likely to be a better solution.
Installing software without admin rights
If possible, install your software using a package manager:
homebrewon OS X.apton Ubuntu (requires admin).conda/mambaotherwise.
conda tips
-
Install miniconda for your system.
-
Add channels in this order:
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
- If you've got an M1 Mac, follow this guide to use Intel packages.
- Never install anything in your base environment.
- To create a new environment, use something like
conda create env -n poppunk python==3.9 poppunk. - Environments are used with
conda activate poppunkand exited withconda deactivate. - Create a new environment for every project or software.
- If it's getting slow at all, use
mambainstead! Use micromamba to get started. - Use
mambafor CI tasks. - You can explicitly define versions to get around many conflicts, in particular changing the python version is not something that will be allowed unless explicitly asked for.
- For help: https://conda-forge.org/docs/user/tipsandtricks.html.
Also note:
- anaconda: package repository and company.
- conda: tool to interact with anaconda repository.
- miniconda: minimal version of the anaconda repostory and conda.
- conda-forge: general infrastructure for creating open-source packages on anaconda.
- bioconda: biology-specific version of conda-forge.
- mamba: a different conda implementation, faster for resolving environments.
What about if your software doesn't have a conda recipe? Consider writing one! Take
a look at an example on bioconda-recipes and copy it to get started.
Exercise 2: Installing a bioinformatics package
Let's try and install bcftools.
To get the code, run:
git clone --recurse-submodules https://github.com/samtools/htslib.git
git clone https://github.com/samtools/bcftools.git
cd bcftools
The basic process for installing C/C++ packages is:
./configure
make
make install
You can also usually run make clean to remove files and start over, if something goes wrong.
Typically, following this process will try and install the software for all users in the system directory /usr/local/bin
which you won't have write access to unless you are root (admin). Usually the workaround is to
set PREFIX to a local directory. Make a directory in your home called software mkdir ~/software and
then run
./configure --prefix=$(realpath ~/software)
make && make install
This will set up some of your directories in ~/software for you. Other directories
you'll see are man/ for manual pages and lib/ for shared objects used by many
different packages (ending .so on Linux and .dylib on OS X).
If there's no --prefix option for the configure script, you can often override
the value in the Makefile by running:
PREFIX=$(realpath ~/software) make
The last resort is to edit the Makefile yourself. You can also change the optimisation
options in the Makefile by editing the CFLAGS (CXXFLAGS if it's a C++ package):
CFLAGS=-O3 -march=native make
Compiles with the top optimisation level and using instructions for your machine.
Try running bcftools -- not found. You'll need to add ~/software to your PATH
variable, which is the list of directories searched by bash/zsh when you type a command:
export PATH=$(realpath ~/software)${PATH:+:${PATH}}
To do this automatically every time you start a shell, add it to your ~/.bashrc or ~/.zshrc file.
Try running make test to test the installation.
To use the bcftools polysomy command requires the GSL library. Install that next. Ideally you'd just use your package manager, but let's do it
from source:
wget https://ftp.gnu.org/gnu/gsl/gsl-latest.tar.gz
tar xf gsl-latest.tar.gz
./configure --prefix=/Users/jlees/software
make -j 4
make install
Adding -j 4 will use four threads to compile, which is useful for larger projects.
Your software/ directory will now has an include/ directory where headers needed
for compiling/making with the library are stored, and a lib/ directory with
libraries needed for running software with the library are stored.
We need to give these to configure:
./configure --prefix=/Users/jlees/software --enable-libgsl CFLAGS=-I/Users/jlees/software/include LDFLAGS=-L/Users/jlees/software/lib
make
Finally, to run bcftools the gsl library also needs to be available at runtime (as opposed
to compile time, which we ensured by passing the paths above). You can
check which libraries are needed and if/where they are found with ldd bcftools (otool -L on OS X):
bcftools:
/usr/lib/libz.1.dylib (compatibility version 1.0.0, current version 1.2.11)
/usr/lib/libSystem.B.dylib (compatibility version 1.0.0, current version 1319.100.3)
/usr/lib/libbz2.1.0.dylib (compatibility version 1.0.0, current version 1.0.8)
/usr/lib/liblzma.5.dylib (compatibility version 6.0.0, current version 6.3.0)
/usr/lib/libcurl.4.dylib (compatibility version 7.0.0, current version 9.0.0)
/Users/jlees/software/lib/libgsl.27.dylib (compatibility version 28.0.0, current version 28.0.0)
/System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib (compatibility version 1.0.0, current version 1.0.0)
So libgsl.dylib and libBLAS.dylib are being found ok. If they aren't being found, you need
to add ~/software/lib to the LD_LIBARY_PATH environment variable like you did with the PATH.
Check that you can run bcftools polysomy.
Troubleshooting jobs and prototyping code
Some tips:
- It's ok to run small jobs on the head node, especially to check you've got the arguments right. Then kill them with
Ctrl-Corkillonce they get going. Avoid running things that use lots of memory (these will likely be killed automatically). You can preface your command withniceto lower its priority. - Find a smaller dataset which replicates your problems, but gets to them without lots of CPU and memory use. Then debug interactively rather than through the job submission system.
- Run a simple or example analysis first, before using your real data or the final set of complex options.
- Add print statements through your code to track where the problem is happening.
- If that doesn't find the issue, use a debugger interactively (see last session). For compiled code giving a segfault, this is usually the way to fix it (or with
valgrind). - If you need to debug on a GPU, be aware this will stop the graphics output.
- Try and checkpoint larger tasks. Save temporary or partial results (in python with a pickle, in R with
saveRDS). Pipeline managers will help with this and let you resume from where things went wrong. - Split your task into smaller jobs. For example, if you have code which parses genomic data into a data frame, runs a regression on this data frame, then plots the results. Split this into three scripts, each of which saves and loads the results from the previous step (using serialisation).
Job arrays
If you have embarrasingly parallel tasks, i.e. those with no shared memory or dependencies, job arrays can be an incredibly convenient and efficient way to manage batching these jobs. Running the same code on multiple input files is a very common example. In some settings, using a job array can give you higher overall priority.
To use an array, add to your bsub command:
bsub -J mapping[1-1000]%200 -o logs/mapping.%J.%I.o -e logs/mapping.%J.%I.e ./wrapper.py
-Jgives the job a name[1-1000]gives the index to run over. This can be more complex e.g.[1,2-20,43], useful for resubmitting failed jobs.%200gives the maximum to run at a time. Typically you don't need this because the scheduler will sort out priority for you, but if, for example, you are using a lot of temporary disk space for each job it may be useful to restrict the total running at any given moment.- We add
%Ito the output file names which is replaced with the index of the job so that every member of the array writes to its own log file.
The key thing with a job array is that an environment variable LSB_JOBINDEX will be
created for each job, which can then be used by your code to set up the process correctly.
Let's work through an example.
Exercise 3: Two-dimensional job array
Let's say I have the following python script which does a stochastic simulation of an infectious disease outbreak using an SIR model and returns the total number of infections at the end of the outbreak:
import numpy as np
def sim_outbreak(N, I, beta, gamma, time, timestep):
S = N - I
R = 0
max_incidence = 0
n_steps = round(time/timestep)
for t in range(n_steps):
infection_prob = 1 - np.exp(-beta * I / N * timestep)
recovery_prob = 1 - np.exp(-gamma * timestep)
number_infections = np.random.binomial(S, infection_prob, 1)
number_recoveries = np.random.binomial(I, recovery_prob, 1)
S += -number_infections
I += number_infections - number_recoveries
R += number_recoveries
return R
def main():
N = 1000
I = 5
time = 200
timestep = 0.1
repeats = 100
beta_start = 0.01
beta_end = 2
beta_steps = 100
gamma_start = 0.01
gamma_end = 2
gamma_steps = 100
for beta in np.linspace(beta_start, beta_end, num=beta_steps):
for gamma in np.linspace(gamma_start, gamma_end, num=gamma_steps):
sum_R = 0
for repeat in range(repeats):
sum_R += sim_outbreak(N, I, beta, gamma, time, timestep)
print(f"{beta} {gamma} {sum_R[0]/repeats}")
if __name__ == "__main__":
main()
The main loop runs a parameter sweep over 100 values of beta and gamma (so \(10^4\) total), and runs 100 repeats (so \(10^6\) total). Each of these parameter combinations takes around 3s to run on my machine, so we'd be expecting about 8 hours total. We could of course use some tricks from last week to improve these, but instead lets use a job array to parallelise.
Do the following:
- Change the
beta_stepsandgamma_stepsto 5 (so we don't all hammer the cluster too hard). - Use
import osandjob_index = os.environ['LSB_JOBINDEX']to get the job index at the start of the main function. - Replace the outer loop over beta with this index, so that the job runs a single beta value, but the full loop over gamma.
- Write an appropriate
bsubcommand to submit this is a job array. Use the-q shortqueue. - Think about how you would collect the results from all of your jobs (using either bash or by modifying the code).
If you have time:
- Also replace the gamma loop, so that each job in the array (of 25 jobs) runs just a single beta and gamma value, but the full repeat loop.
- Change the print statement so it's easier to collect your results at the end.
It may help to review the section on strides from the previous session when you are thinking about how to map a 1D index into two dimensions.
One other note: here we are using random number generators across multiple processes. If they have the same seed, which may be the default, they will produce identical results and likely be invalid repeats. Even if they are set to different seeds (either by using the job index or by using the system clock), after some time they will likely become correlated, which may also cause problems. See Figure 3 in this paper for how to fix this.
Reproducibility on the HPC
Tips based on what has worked for me:
- Keeping track of your command history goes a long way. Set up your history options with unlimited scrollback and timestamps.
- Search through history in the following order:
- Type the start of your command, then press up.
- Use Ctrl-r to search for part of your command.
- Use
history | grepfor more complex searches. e.g.history | grep sir.py | grep bsubwould find when thesir.pyscript was submitted with bsub. You can use-Cto add context and see other commands. Or usehistory | lessand the search function to find the sequence of commands you used.
- Be careful with editing files in a text editor on the cluster -- this command cannot just be rerun and it won't be clear what you did.
- Use git for everything you write, and commit often.
- For larger sets of jobs, particularly which you will run repeatedly or expect some failures, it may be worth using pipeline software (snakemake, nextflow) to connect everything together. Ususally they can handle the
bsub/SLURM submission for you. - Keep your code and data separate.
- Don't modify your input data, instead make intermediate files.
- Don't overwrite files unless you have to. Regularly clean and tidy files you are working on.
- Call your files and directories sensible names and write a
READMEif they get complicated. - Think about your main user: future you. Will you be able to come back to this in a month's time? Three year's time?
- Use
-oand-ewith all jobs, and write tologs/folders. You can easilytarthese once you are done. - Make plots on your local machine (with code that can be run through without error or needing the current environment). Transfer smaller results files from the HPC to make your plots.
- Use conda environments to make your software versions consistent between runs. Use
conda listto get your software and versions when you write papers.